|
SECOND PLACE WINNER!
Best of Naturopathic Medicine 2013
Page 1, 2, 3
Background and Innovation
"There are no naturally occurring beta blockers," Dr. Milner often says in reference to the use of beta-1 adrenergic receptor blockers (beta blockers) in the treatment of arrhythmias and in control of the heart rate. "Nature doesn't block receptors," Milner asserts. "It augments antagonist pathways. Acetylcholine is 'nature's beta blocker.'"
There are moments in a physician's career when he sees the biochemistry of a disease process in a whole new way. Beginning in 2008, Dr. Milner revisited his study of the autonomic nervous system (ANS) control of heart rate; he considered the actions of beta blockers which inhibit sympathetic ANS stimulation of the heart. He considered acetylcholine (ACh), the neurotransmitter of the parasympathetic ANS, and its action on heart rate inhibition. He then developed an approach for using oral supplements to augment ACh production and release. This evolved into the Milner Acetylcholine Protocol (MAP) for management of cardiac dysrhythmias.1
Biochemical Plausibility
The biochemical plausibility of the MAP is based on the ACh inhibitory effect on cardiac rate and dysrhythmias through several mechanisms. The sinoatrial (SA) node, the normal pacemaker of the heart, receives input from the 10th cranial nerve (CN X), the vagus nerve, via ACh. That effect on the SA node inhibits automaticity and slows the heart rate. This is achieved by hyperpolarization of the cells through increased potassium permeability, resulting in slower spontaneous depolarization.2
The atrioventricular (AV) node of the heart also receives input from the vagus nerve via ACh. The effect slows or blocks conduction through the node. Again, this is achieved through a prolonged refractory period that results from increased cell permeability to potassium, which increases the phase 4 portion of the cardiac action potential.
The ventricles of the heart are also affected by ACh, though the vagal innervation of the ventricles is sparse. This occurs by virtue of the antagonistic relationship between the sympathetic and parasympathetic halves of the ANS.
Ventricular myocardium is heavily innervated by the sympathetic ANS. Norepinephrine (NE) is the dominant neurotransmitter which agonizes the beta-1 adrenergic receptors of the heart; it increases automaticity and contractility by increasing the permeability of the cells to calcium. Release of ACh in the supraventricular myocardium and from the few parasympathetic fibers in the ventricles inhibits the release of NE from sympathetic fibers. That NE inhibition prevents increased automaticity by the sympathetic ANS.3
ACh in the human body is synthesized from acetyl-coenzyme A (acetyl-CoA) and choline in a reaction catalyzed by the enzyme choline acetyl transferase (CAT; Figure 1).
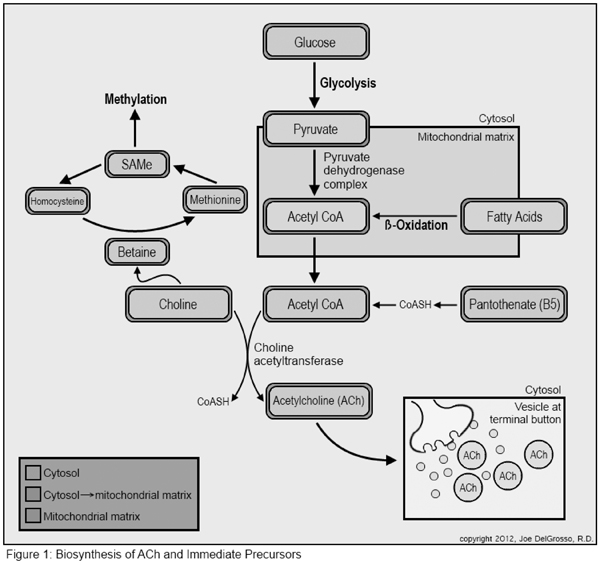
Acetyl-CoA comes from any of several sources, including dehydrogenation of pyruvate, end products of fatty acid metabolism, or production of coenzyme A (CoA) from vitamin B5 which is then acetylated before interacting with choline.
Pyruvate is the end product of aerobic glycolysis. It can also be converted from lactate, the end product of anaerobic glycolysis. Pyruvate can then be converted to acetyl-CoA through the action of the pyruvate dehydrogenase complex. Acetyl-CoA then has several potential fates, all of which demand the transfer of acyl groups, including participation in the Krebs cycle, in fatty acid synthesis, in the production of ACh, and others.4
Pantothenic acid (PA), vitamin B5, is the initial substrate for de novoCoA synthesis. This five-step process requires both pantothenate (ionized PA) and the sulfur containing amino acid cysteine. Pantethine is a derivative of PA and is the functional component of CoA in the carrying of acyl groups. Pantethine is composed of two pantethine molecules joined by a disulfide bond.5
Choline is an essential nutrient. Humans reutilize choline and synthesize it de novo, but in quantities insufficient to meet metabolic demand. Therefore, an adequate intake of choline is necessary for health.6 The established adequate intake (AI) in adult men is 550 mg/day and in adult women is 425 mg/day unless pregnant (450 mg/d) or breast-feeding (550 mg/d). Eggs are the richest dietary sources of choline, followed by animal protein sources, leaving vegans and several other groups at routinely inadequate daily intakes.7–10 A list of choline content in foods can be found in the Micronutrient Information Center article on choline from the Linus Pauling Institute, Oregon State University.7
Choline is an essential substrate in the synthesis of the neurotransmitter ACh, but is also vital in several other bodily processes. Choline is necessary: as a component of cell membranes, in cell signaling molecules, in lipid metabolism and transportation, and as a major source of methyl groups. Methylation reactions turn body processes on and off and activate and inactivate metabolic intermediates.7
Choline deficient states may play a part in diseases and conditions including liver disease and fatty liver infiltration, neural tube defects, methylation defects, hypertension, atherosclerosis, neurological disorders, inflammatory disorders, and cancer.7 Population research has demonstrated that many groups do not achieve their AI of choline.6,8–11 For instance, there is evidence that only 2% of postmenopausal women reach their choline AI on a daily basis.11
Research has established that genetic polymorphisms which alter choline or folate metabolism may increase an individual's dietary requirement for choline. For instance, when 5-methyltetrahydrofolate (5-MTHF) is deficient due to a polymorphism (or other reasons), the body depends on betaine from choline for the conversion of homocysteine to methionine.12 Methionine is then converted to S-adenosyl-methionine (SAMe), the major methyl donor for one-carbon transfers in the body.13 When choline is required as a methyl donor it is shunted away from other biochemical processes. Therefore, greater demand for dietary choline may be prevalent in the population based on genetic polymorphisms that are prevalent.14–16
Therapeutic Plausibility
The therapeutic plausibility of utilizing the ACh pathway in the treatment of cardiac dysrhythmias through supplementing oral precursors requires both that (1) oral supplementation be able to raise ACh levels in the neurons; and (2) those increases demonstrate clinical effects.
It is well established that the amount of ACh in the central nervous system is directly affected by the oral intake of choline. Studies and reviews which date back to the late 1970s establish that the synthesis of ACh in the brain (CNS) responds to the availability of choline in the blood and that the ability of neurons to make and release neurotransmitters, including ACh, depends directly on the concentration of amino acids and choline in the blood.17,18
Excellent research has been produced by Marty Hinz, MD, and his groups over the recent decades substantiating that oral supplementation of amino acid precursors can raise endogenous neurotransmitter levels. His work pertains primarily to serotonin and catecholamines, but is applicable to ACh via analogous mechanisms. More importantly, the work of Hinz et al. has established that oral precursor dosing leads to demonstrable clinical effects and improved patient outcomes.19–22
The half-life of ACh is very short at 2 minutes. To ensure fidelity in neuromuscular transmission, the body further enhances this breakdown of ACh enzymatically through acetylcholine esterase enzymes. While ACh is reclaimed by reuptake at the synaptic cleft, augmenting production and release to the extent necessary to inhibit cardiac automaticity will require the persistent availability of substrates for ACh production, a problem that we will need to address.
While we can find no evidence in the medical literature for the use of choline to address cardiac dysrhythmias, we can establish that the biochemical action of ACh exerts control over the cardiac rate and depolarization. The fact that supplementing oral choline raises levels of ACh in the CNS is long established in the medical literature, as cited.17,18 Therefore, the biochemical and therapeutic plausibility for the choline aspect of MAP is present.
Pantethine has been reported to have antiarrhythmic action on experimental models of cardiac arrhythmias. In a study of a dog model of hypoxic cardiac perfusion, administration of pantethine was shown to significantly prolong the cardiac action potential and refractory period.23 While the mechanism of that effect was not described, it is noteworthy that this is the same action which ACh has on the supraventricular myocardial cells, as described above.
Research on rate models of cardiac function has demonstrated that the intravenous (IV) administration of CoA results in a transient decrease in the heart rate. These studies make it appear that it is the adenosine portion of the CoA molecule which may be responsible for the decrease in heart rate rather than the pantethine, pantothenic acid, or cysteine constituents of the molecule.24
The accumulation of long chain acyl fatty acid intermediates promotes cardiac arrhythmias. This has been demonstrated in several respects. Cardiac arrhythmia may be the first clinical presentation in children with fatty acid oxidation disorders.25 Experimental models have demonstrated that buildup of acylcarnitines and other intermediates promote dysregulation of calcium resulting in dysrhythmias.26 Ischemic injury in myocardial infarction has also been associated with the buildup of these intermediates promoting arrhythmogenesis. Meanwhile, shorter chain metabolites such as acetyl-CoA and palmitoyl-CoA do not promote arrhythmogenesis.26,27
The potential for statin drugs (HMG-CoA reductase inhibitors) to exert an antiarrhythmic effect on the heart has been an area of research interest. Statins reduce recurrences of supraventricular and ventricular arrhythmias in patients with and without coronary artery disease. Several mechanisms have been proposed for this antiarrhythmic effect, yet none have been proved. Hypotheses include: lowering of LDL cholesterol, improved endothelial function, stabilization of atherosclerosis, anti-inflammatory effects, modulation of membrane ion flux, and improvement of autonomic function.28,29 Experiments using other antilipidemic agents have not shown similar antiarrhythmic effects; therefore something unique about statins must give them their antiarrhythmic properties, rather than their LDL lowering or atherosclerosis stabilizing effects.
Statin drugs inhibit the effect of the HMG-CoA reductase enzyme, leading to an upstream increase in metabolic substrates, especially CoA. It may be that buildup of CoA is responsible, in whole or in part, for the antiarrhythmic effects of statins or for an improvement in autonomic function.
Therapeutic plausibility for the effect of CoA/pantethine/PA on cardiac dysrhythmias is present in the medical literature. Since biochemical and therapeutic plausibility exists for use of either PA/pantethine or choline singly, the potential exists for their combined effect to be greater than the sum of its parts. This is the principle of the MAP.
Page 1, 2, 3
|




